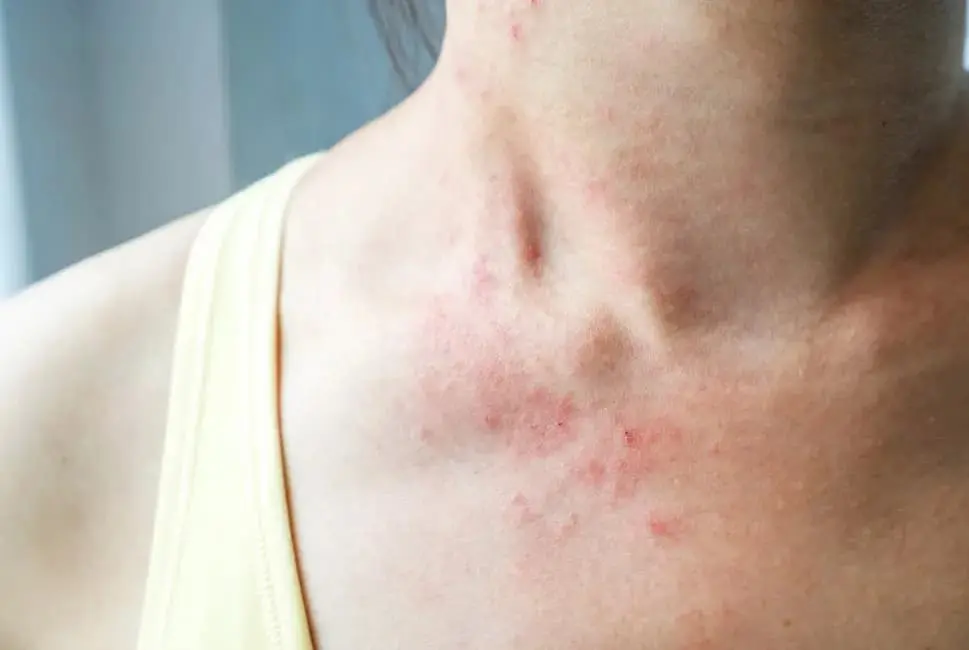
1. Introduzione alla Sindrome di Stevens-Johnson
La Sindrome di Stevens-Johnson è una malattia dermatologica acuta, a esordio improvviso, che si manifesta come una grave reazione di ipersensibilità, prevalentemente di origine farmacologica. Si tratta di una condizione rara ma estremamente seria, con un potenziale letale se non trattata tempestivamente. La sua rilevanza medica è legata sia alla difficoltà nella diagnosi precoce che all’impatto devastante sullo stato generale di salute del paziente.
Il riconoscimento precoce dei sintomi, l’identificazione della causa scatenante (spesso un farmaco) e l’inizio immediato di un trattamento di supporto possono fare la differenza tra un recupero efficace e complicanze potenzialmente irreversibili. In questo articolo analizziamo in modo approfondito cos’è la SJS, come riconoscerla, quali sono i fattori scatenanti, le modalità diagnostiche, le opzioni terapeutiche attuali e le strategie di prevenzione.
2. Cos’è la Sindrome di Stevens-Johnson?
La Sindrome di Stevens-Johnson è una patologia dermatologica immunomediata che rientra nello spettro delle dermatosi bollose acute. Viene classificata come una delle più gravi reazioni avverse ai farmaci (ADR – Adverse Drug Reactions). È caratterizzata dalla necrosi massiva delle cellule dell’epidermide, che si separa dal derma sottostante, provocando desquamazione cutanea e ulcerazioni dolorose delle mucose.
Si stima che colpisca circa 1-6 persone su un milione all’anno, con una maggiore incidenza nei pazienti immunocompromessi (es. HIV positivi). Il quadro clinico della SJS è sovrapponibile alla Necrolisi Epidermica Tossica (TEN), ma la differenza principale risiede nell’estensione del danno cutaneo:
-
SJS: <10% della superficie corporea interessata.
-
SJS/TEN sovrapposta: 10-30%.
-
TEN: >30%.
La separazione epidermica porta a gravi perdite di liquidi, rischio di sepsi, complicanze oculari e danni permanenti alla pelle e alle mucose. La SJS è una vera emergenza dermatologica e sistemica.
3. Identificare i Sintomi della Sindrome di Stevens-Johnson
I sintomi della SJS possono essere ingannevoli nelle fasi iniziali, poiché mimano quelli di una comune infezione virale. Tuttavia, la progressione rapida verso manifestazioni cutanee ed epiteliali drammatiche richiede una diagnosi differenziale rapida.
Fase iniziale (prodromica)
-
Febbre alta persistente.
-
Faringodinia (mal di gola).
-
Brividi.
-
Affaticamento intenso.
-
Artralgie e mialgie.
-
Congiuntivite oculare (primo segnale mucosale).
Fase acuta
-
Eruzione maculo-papulare che evolve in bolle e vescicole, spesso dolorose.
-
Coinvolgimento delle mucose (90-100% dei casi): ulcere orali, lesioni oculari, genitali e anali.
-
Nikolsky positivo: la pelle si distacca al minimo sfregamento.
-
Desquamazione massiva (simile a un’ustione di secondo grado).
Il coinvolgimento oculare può determinare cheratocongiuntivite, ulcerazioni corneali, secchezza oculare cronica o addirittura cecità. I sintomi peggiorano nell’arco di 24-72 ore e il quadro clinico richiede intervento ospedaliero immediato.
4. Comprendere le Cause e i Fattori di Rischio della Sindrome di Stevens-Johnson
Principali cause
La causa più comune della SJS è una reazione avversa a farmaci, con un meccanismo di ipersensibilità ritardata. I farmaci più frequentemente implicati includono:
-
Antibiotici: sulfamidici (es. cotrimossazolo), penicilline, cefalosporine.
-
Anticonvulsivanti: carbamazepina, lamotrigina, fenitoina.
-
Allopurinolo.
-
FANS: ibuprofene, naprossene, diclofenac.
-
Antiretrovirali e altri farmaci oncologici.
Infezioni correlate
-
Virus: Herpes simplex, Epstein-Barr, HIV.
-
Batteri: Mycoplasma pneumoniae.
-
Vaccinazioni (molto raramente).
Fattori genetici
-
La presenza di alleli HLA specifici può predisporre alla SJS. Esempio: HLA-B1502* è fortemente associato alla SJS indotta da carbamazepina nei soggetti asiatici.
-
Mutazioni nel gene CYP450 possono alterare il metabolismo dei farmaci.
Altri fattori di rischio
-
Immunosoppressione (es. HIV, trapianti).
-
Precedenti reazioni cutanee.
-
Età avanzata.
-
Polifarmacoterapia.
5. Diagnosi e Opzioni di Trattamento della Sindrome di Stevens-Johnson
Diagnosi
-
Esame clinico accurato dei segni cutanei e mucosali.
-
Anamnesi farmacologica dettagliata (ultimo mese).
-
Biopsia cutanea: evidenzia necrosi dell’epidermide e infiltrato linfocitario.
-
Test ematici: emocromo, elettroliti, enzimi epatici e renali.
-
Esami colturali: per identificare infezioni secondarie.
Trattamento
Il trattamento è multidisciplinare e va eseguito in ambienti specializzati:
Sospensione del farmaco scatenante
Essenziale entro le prime 24-48 ore per ridurre la progressione.
Supporto intensivo:
-
Reidratazione per via endovenosa.
-
Correzione degli elettroliti.
-
Terapia nutrizionale (enterale o parenterale).
-
Monitoraggio della pressione arteriosa e della diuresi.
Gestione delle lesioni cutanee:
-
Uso di garze non aderenti.
-
Antisettici topici (es. clorexidina).
-
Terapia del dolore.
Prevenzione delle infezioni:
-
Antibiotici sistemici solo se infezione confermata.
-
Isolamento in camera sterile (se disponibile).
Trattamenti sistemici adiuvanti:
-
Corticosteroidi (controversi, ma usati in fase iniziale).
-
Immunoglobuline EV (IVIG).
-
Ciclosporina o altri immunosoppressori in casi gravi.
Gestione oculistica precoce:
-
Colliri antibiotici e lubrificanti.
-
Intervento oftalmologico specialistico per evitare danni permanenti.
6. Prevenzione e Gestione a Lungo Termine della Sindrome di Stevens-Johnson
Prevenzione
-
Evitare i farmaci noti per causare SJS nei soggetti predisposti.
-
Utilizzo di test genetici HLA in soggetti ad alto rischio (es. prima della prescrizione di carbamazepina in asiatici).
-
Educazione del paziente su sintomi precoci e auto-monitoraggio.
Follow-up e gestione delle sequele
-
Cicatrici cutanee e aderenze mucosali che possono compromettere la qualità della vita.
-
Complicanze oculari: secchezza cronica, opacità corneale, cecità.
-
Dolore cronico e disfunzioni sessuali (in caso di coinvolgimento genitale).
-
Supporto psicologico: sindrome da stress post-traumatico, ansia, depressione.
-
Allergologia: per valutare le future tolleranze farmacologiche.
Prognosi
Il tasso di mortalità per la SJS varia tra il 5% e il 15%, mentre per la TEN può superare il 30%. Un recupero completo è possibile, ma molti pazienti riportano esiti permanenti.
7. Domande Frequenti sulla Sindrome di Stevens-Johnson (FAQ)
Cos’è la Sindrome di Stevens-Johnson?
La Sindrome di Stevens-Johnson è una reazione avversa rara ma grave che colpisce la pelle e le mucose, causata principalmente da farmaci. Si tratta di una forma acuta di necrolisi epidermica che porta al distacco dell’epidermide e alla formazione di vesciche e ulcere dolorose. La SJS può coinvolgere la bocca, gli occhi, i genitali e può evolvere rapidamente in una condizione potenzialmente letale se non trattata in tempo. È considerata un’emergenza medica che richiede ricovero ospedaliero immediato.
Quali farmaci possono causare la Sindrome di Stevens-Johnson?
Diversi farmaci possono scatenare la SJS, in particolare:
-
Antibiotici: come sulfamidici (es. cotrimossazolo), penicilline, cefalosporine.
-
Anticonvulsivanti: come carbamazepina, lamotrigina, fenitoina.
-
Allopurinolo: usato per la cura della gotta.
-
FANS (farmaci antinfiammatori non steroidei): come ibuprofene e naprossene.
-
Antivirali e farmaci oncologici in pazienti immunocompromessi.
La SJS può manifestarsi anche 1-3 settimane dopo l’inizio del trattamento, rendendo difficile l’identificazione del farmaco responsabile se non si analizza attentamente la storia clinica recente.
Quali sono i primi sintomi della Sindrome di Stevens-Johnson?
I sintomi iniziali sono aspecifici e possono essere confusi con una comune influenza:
-
Febbre alta e persistente.
-
Mal di gola, tosse e malessere generale.
-
Dolori muscolari e articolari.
-
Sensazione di bruciore agli occhi.
Nell’arco di poche ore o giorni compaiono:
-
Eruzioni cutanee che evolvono in vesciche dolorose.
-
Lesioni alle mucose (bocca, occhi, genitali).
-
Desquamazione della pelle, in particolare sul volto, tronco e arti.
-
Difficoltà a deglutire, fotofobia e bruciore oculare.
L’aggravamento rapido dei sintomi richiede un intervento medico urgente.
La Sindrome di Stevens-Johnson è contagiosa?
No, la SJS non è una malattia contagiosa. Non si trasmette da persona a persona, in quanto si tratta di una reazione del sistema immunitario a una sostanza (di solito un farmaco o un’infezione). Tuttavia, chi ha avuto una SJS in passato è a rischio di una recidiva se riassume lo stesso farmaco o un farmaco simile.
Come viene diagnosticata la Sindrome di Stevens-Johnson?
La diagnosi si basa su:
-
Esame clinico: valutazione dell’aspetto delle lesioni cutanee e mucosali.
-
Anamnesi farmacologica: identificazione di farmaci assunti di recente.
-
Biopsia cutanea: conferma istologica con evidenza di necrosi epidermica.
-
Esami del sangue: per monitorare lo stato generale del paziente e verificare eventuali complicanze.
-
Esami colturali: per rilevare eventuali infezioni secondarie.
La tempestività nella diagnosi è essenziale per interrompere il farmaco sospetto e iniziare il trattamento di supporto.
Qual è il trattamento per la Sindrome di Stevens-Johnson?
Non esiste una cura specifica, ma il trattamento si basa su:
-
Interruzione immediata del farmaco sospetto.
-
Ospedalizzazione, preferibilmente in centri ustioni o terapia intensiva.
-
Supporto vitale: idratazione EV, bilancio elettrolitico, nutrizione.
-
Cura delle lesioni cutanee e delle mucose.
-
Prevenzione delle infezioni con misure igieniche e, se necessario, antibiotici.
-
Terapia del dolore.
-
Supporto oftalmologico per prevenire danni irreversibili agli occhi.
-
In casi selezionati: corticosteroidi sistemici, immunoglobuline EV, o immunosoppressori.
Il recupero può richiedere settimane e la riabilitazione deve essere multidisciplinare.
Esiste un modo per prevenire la Sindrome di Stevens-Johnson?
Prevenire la SJS non è sempre possibile, ma alcune precauzioni riducono il rischio:
-
Evitare farmaci già noti per aver causato reazioni allergiche.
-
Informare sempre il medico di reazioni avverse precedenti.
-
Nei soggetti a rischio (es. asiatici), è possibile fare test genetici HLA prima della somministrazione di farmaci ad alto rischio (es. carbamazepina).
-
Limitare l’uso di farmaci non necessari o assunti in modo autonomo.
-
Monitorare attentamente l’introduzione di nuovi farmaci, soprattutto nei primi 30 giorni.
Quali sono le prospettive a lungo termine per chi ha avuto la Sindrome di Stevens-Johnson?
La prognosi varia in base alla gravità del caso e alla rapidità dell’intervento. Possibili esiti a lungo termine includono:
-
Cicatrici cutanee e iperpigmentazioni.
-
Adesioni mucosali a livello genitale o orale.
-
Complicanze oculari: secchezza cronica, cheratiti, cecità.
-
Sensibilità ai farmaci futuri.
-
Impatto psicologico: ansia, depressione, sindrome da stress post-traumatico.
Alcuni pazienti si riprendono completamente, mentre altri necessitano di cure a lungo termine. È fondamentale un follow-up medico regolare e il supporto di specialisti (oculista, dermatologo, allergologo, psicologo).
Quanto tempo impiega la Sindrome di Stevens-Johnson a manifestarsi dopo l’assunzione di un farmaco?
La SJS può comparire da 1 a 3 settimane dopo l’inizio di un nuovo farmaco. Tuttavia, in pazienti che hanno già avuto una reazione precedente, la manifestazione può essere molto più rapida, anche dopo poche ore. Per questo motivo, è fondamentale segnalare sempre al medico eventuali precedenti reazioni allergiche a farmaci.
Qual è la differenza tra Sindrome di Stevens-Johnson e Necrolisi Epidermica Tossica (TEN)?
La differenza principale tra SJS e TEN sta nell’estensione del danno cutaneo:
-
SJS: coinvolge meno del 10% della superficie corporea.
-
TEN: coinvolge oltre il 30%.
-
SJS/TEN sovrapposta: coinvolgimento tra il 10% e il 30%.
Clinicamente, i sintomi sono simili, ma la TEN ha una prognosi peggiore e un rischio di mortalità molto più alto.
Posso sviluppare la Sindrome di Stevens-Johnson più di una volta?
Sì. Chi ha avuto una SJS in passato ha un rischio elevato di recidiva se viene riesposto al farmaco responsabile o a farmaci simili. È per questo che i pazienti dovrebbero sempre:
-
Portare con sé un braccialetto di allerta medica.
-
Includere l’informazione nella propria cartella clinica elettronica.
-
Informare tutti i professionisti sanitari coinvolti nella cura.
Quali esami genetici possono essere utili per prevenire la SJS?
In alcune popolazioni, sono disponibili test genetici per identificare la predisposizione alla SJS, in particolare:
-
HLA-B*1502: test utile nei pazienti di origine asiatica prima dell’uso di carbamazepina.
-
HLA-A*3101: associato a rischio aumentato con altri anticonvulsivanti.
Questi test vengono generalmente eseguiti solo nei soggetti a rischio o prima di iniziare farmaci specifici ad alto potenziale di reazione.
Un’infezione può scatenare la Sindrome di Stevens-Johnson?
Sì. Anche se i farmaci rappresentano la causa più comune, alcune infezioni virali o batteriche possono innescare la SJS, specialmente:
-
Mycoplasma pneumoniae (frequente nei bambini).
-
Virus Herpes simplex.
-
HIV.
In questi casi, il trattamento si concentra sull’eliminazione dell’agente infettivo e sul supporto generale del paziente.
È pericoloso usare rimedi naturali o erboristici se ho avuto la SJS?
Sì. Anche alcuni prodotti erboristici o integratori naturali possono causare reazioni immunitarie gravi in soggetti predisposti, soprattutto se combinati con farmaci. Non è consigliabile assumere nessun rimedio naturale senza averne parlato con il medico.
I bambini possono sviluppare la Sindrome di Stevens-Johnson?
Sì. La SJS può colpire anche i bambini, sebbene sia più rara rispetto agli adulti. Nei bambini, spesso la causa è legata a infezioni (es. Mycoplasma pneumoniae) più che a farmaci. La gravità è simile a quella degli adulti e il trattamento richiede ugualmente ospedalizzazione.
La SJS può causare infertilità?
In alcuni casi gravi in cui vengono coinvolti i genitali, lesioni ulcerative profonde e aderenze possono interferire con le normali funzioni sessuali o riproduttive, specialmente se non trattate tempestivamente. È importante un follow-up ginecologico o andrologico per prevenire complicazioni permanenti.
Dopo quanto tempo dalla guarigione posso tornare alla vita normale?
Il ritorno alla normalità dipende dalla gravità del caso:
-
Nei casi lievi/moderati: alcune settimane per il recupero completo.
-
Nei casi gravi con lesioni estese o complicanze oculari: possono essere necessari mesi di riabilitazione.
È importante sottoporsi a controlli regolari e seguire indicazioni mediche personalizzate per una ripresa sicura e duratura.
Esiste un registro per le reazioni avverse gravi come la SJS?
Sì. In molti paesi esistono registri o sistemi di farmacovigilanza dove i medici possono segnalare casi di reazioni avverse gravi, tra cui la SJS. In Italia, la segnalazione può essere effettuata tramite il sito dell’AIFA (Agenzia Italiana del Farmaco). Questo aiuta a migliorare la sicurezza dei farmaci sul mercato e a tutelare i pazienti.
Fonti dell’articolo:
American Academy of Dermatology (AAD).
Tutte le informazioni contenute in questo articolo sono fornite a solo scopo informativo, in nessun caso costituiscono la formulazione di una diagnosi o la prescrizione di un trattamento, e non intendono e non devono in alcun modo sostituire il rapporto diretto medico-paziente o la visita specialistica. Si raccomanda di chiedere sempre il parere del proprio medico curante e/o di specialisti riguardo qualsiasi indicazione riportata. Come specificato in ogni articolo se si hanno dubbi o quesiti sull’uso di un farmaco è necessario contattare il proprio medico.
